
こんにちは、risachicaです。
今年2024年のノーベル生理学・医学賞受賞はゲイリー・ラブカン教授の「マイクロRNAと遺伝子調節における役割の解明」でしたね!
ノーベル化学賞とともに、まさに今の製薬業界の開発の新技術の実現を表している感じで、胸熱でした。
miRNA技術とは:
短いRNA配列をmRNAの非翻訳領域に結合させ、蛋白質の生産をコントロールする技術です。疾患の原因になる蛋白質の発現を抑制、あるいは補充することで、疾患を改善させます。現在あるiRNA技術を用いた薬剤の対象疾患として、癌、心疾患、免疫・神経炎症疾患、感染症、代謝性疾患等があります。今現在積極的に開発がすすめられており、今後新規に薬剤が出てくるであろうアツイ領域であることは間違いないと思います!!
研究開発をしていた頃にこれが実現したらすごいことになる…とRNA技術の可能性を感じてワクワクしていた私は
miRNA技術がノーベル賞という最高峰の賞を受賞するのを現実に見て本当に実用化に向かっていくと思えて、とてもうれしいです。
今はAI技術もそうですが、技術の発展に伴いすべてのことが一気に劇的に変化しているので、これからもどんどんすごいことが本当に起こる時代にいるのだと思うと、ワクワクしますね。
興味深いタイムリーなトピックなので書いてしまいましたが
今日は、薬の開発に伴う安全性報告がいつ?どのような報告をする必要があるか?について書いていきたいと思います。
なぜ副作用報告が必要なのか?
まず、薬剤の開発をして、その薬を販売してよいと国から承認を得るために
有効性と同時に安全性の情報についても収集する義務があります。
そのことは医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(通称、薬機法)に規定されています。
第六十八条の十
(副作用等の報告)
第六十八条の十医薬品、医薬部外品、化粧品、医療機器若しくは再生医療等製品の製造販売業者又は外国特例承認取得者は、その製造販売をし、又は第十九条の二、第二十三条の二の十七若しくは第二十三条の三十七の承認を受けた医薬品、医薬部外品、化粧品、医療機器又は再生医療等製品について、当該品目の副作用その他の事由によるものと疑われる疾病、障害又は死亡の発生、当該品目の使用によるものと疑われる感染症の発生その他の医薬品、医薬部外品、化粧品、医療機器又は再生医療等製品の有効性及び安全性に関する事項で厚生労働省令で定めるものを知つたときは、その旨を厚生労働省令で定めるところにより厚生労働大臣に報告しなければならない。
第六十八条の十第1項には製造販売業者の副作用報告義務、
第六十八条の十第2項には医療関係者の副作用報告義務、
第六十八条の十第3項にはPMDAの救済給付を申請者の副作用についてMHLWへの報告義務が書かれています。
これら大元の法律をもとに、GPSP省令、GVP省令等でブレイクダウンして書かれてます。
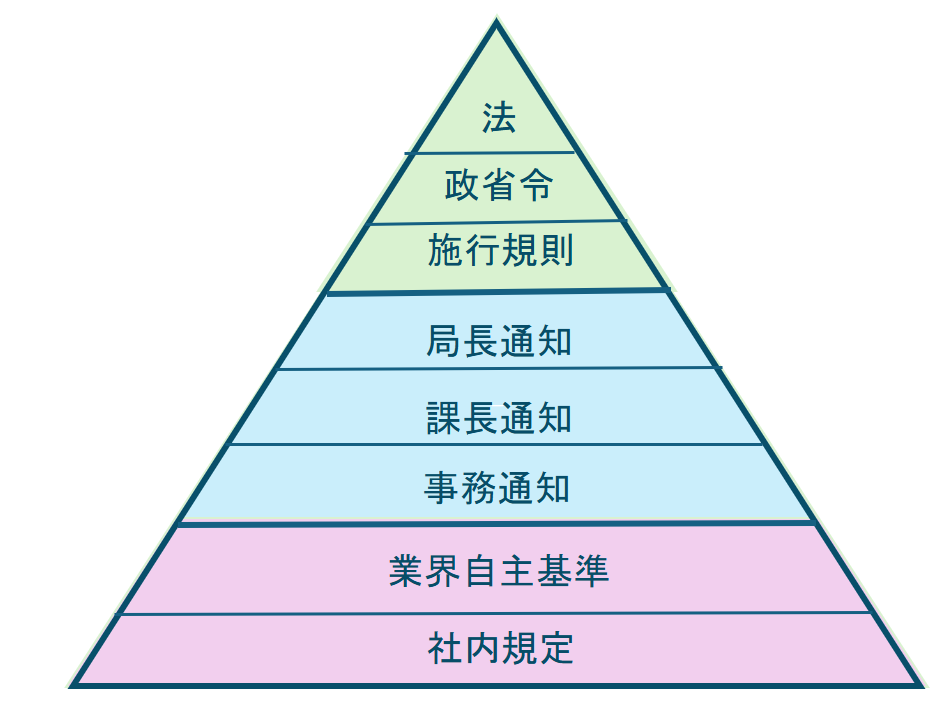
法規制と行政指導
現場での安全性情報報告の細かいやり方について
いつから安全性情報を収集する必要があるか?
薬の開発に伴い安全性情報を収集する義務があることは分かりましたが、ではいつから安全性収集する義務が生じるか?です。
先に答えを書くと、初回治験届(CTN: )を提出したら です。
CTNを提出して受理されたらすぐに安全性情報を収集義務が生じるため、
薬事や臨床などと連絡をとり、いつ提出するのかしっかり連携していく必要があります。
ここで注意ですが、提出日が安全性情報収集開始日であるのは日本だけであり、諸外国はCTN承認日が安全性情報収集開始の日付となるのが通常です。
なのでグローバルの臨床と緊密に協働し、「日本のCTN提出日にあわせて、報告が必要と思われる安全性情報をわたしてくださいね!」とお願いしていく必要があります。
大企業の場合は日本への症例情報のroutingが自動化していますが
小規模ベンチャーで症例対応を委託先の安全性データベースで行っている場合等は、マニュアルで行います。
また、どちらかといえば、薬事の人はCTD等の書類提出に集中しているため、
CTN提出後すぐに収集義務が生じるので、提出・受理されたら遅滞なく知らせてもらえるよう
薬事部の動向を注視して、情報を渡してもらえるよう、PVから働きかけていかないといけないです。
いつまで安全性情報の収集が必要か?
ここまで安全性情報収集の開始について、書いてきました。
次に、治験を行っている間、いつまで治験の安全性情報収集するのが必要かですが、
開発が中止されるまで、あるいは薬剤が承認されるまで です。
「E2B(R3) 実装ガイドに対応した市販後副作用等報告及び治験副作用等報告について」の一部改正について (wakayama.lg.jp)(令 和 2年8月 31 日 発行)
ウ.報告義務期間の取扱い
(ア)報告義務期間
① 被験薬に係る報告義務期間は、当該被験薬について、初めて届書を提出した日か
ら、承認を取得するまで又は開発中止届書を提出するまでのいずれかの期間とする。
なお、治験計画届書の提出を要しない場合は、当該被験薬の治験実施計画書に記載
している実施期間の開始日から、承認を取得するまで又は開発を中止する旨を機構
審査マネジメント部審査企画課に書面(様式自由)により申し出るまでのいずれか
の期間とする。
② 被験薬以外の治験使用薬に係る報告義務期間は、当該治験使用薬を用いる治験に
係る届書を提出した日から、当該治験に係る治験終了届書を提出するまで、当該治
験における被験薬が承認を取得するまで又は当該被験薬の開発中止届書を提出する
までのいずれかの期間とする。なお、治験計画届書の提出を要しない場合は、当該
治験使用薬を用いる治験実施計画書に記載している実施期間の開始日から、終了日
まで、当該治験における被験薬が承認を取得するまで又は当該被験薬の開発を中止
する旨を機構審査マネジメント部審査企画課に書面(様式自由)により申し出るま
でのいずれかの期間とする。
たとえば、残念ながら薬剤成分の開発が何らかの理由によって中止されることとなった場合、開発中止届が提出される日まで、安全性情報の収集義務があります。
治験中止の場合は、安全性情報収集義務が継続します。
そして、この中止の理由が予算の問題や、薬剤の収益性等ビジネス上の判断によるものでなく、安全上の問題である場合
治験の中止は、緊急の措置として別に措置報告として15日以内に報告する必要があります。
(※措置報告については、将来的にまた記事を書くつもりです)
また、無事に承認された場合は
その後は市販後としての報告が必要となります。
このへんのそれぞれの入力事項については通知によって定義されているため、現場では注意深いハンドリングが必要になってきます…
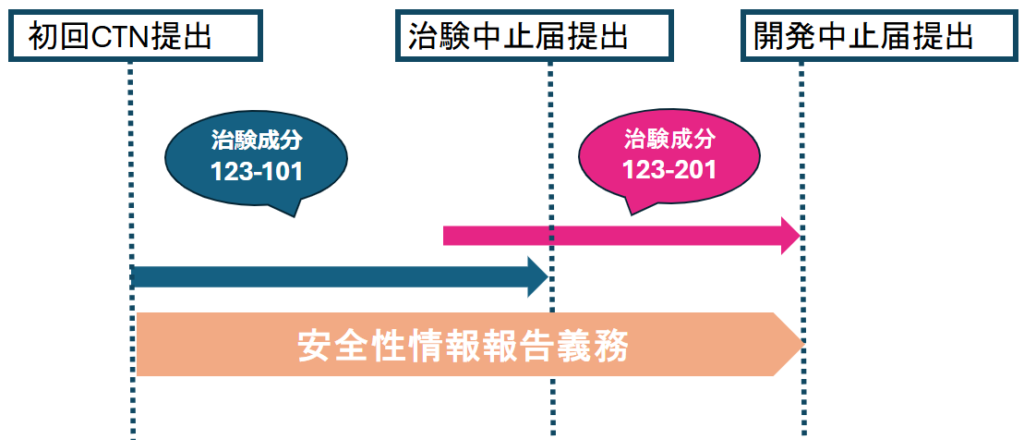
治験中止と開発中止とは?
薬剤の治験の中止には、治験の中止と開発の中止があります。
どう違うのか??
- 治験の中止:その成分を用いた、その治験を中止する。
※(例)成分記号123-201 (phase 2)試験を中止。その成分を用いた他の治験は継続している可能性がある
→措置報告が必要 - 開発の中止:その成分の薬剤としての開発自体を中止する。
この場合は、措置報告の要件(6)、(5)にそれぞれ該当し、①安全性の問題が生じた時、②治験中止届を提出した時の2つの時点で措置報告が必要となります。
下記、安全性情報収集の届出とそれに伴う対応を時系列で示したイメージです。
留保届と留保解除申請書
留保届/留保解除申請書の提出は、安全性情報提出の停止/再開とダイレクトにつながっています。
治験中止後にも安全性情報については報告する必要がありますが、
留保届を提出することで安全性報告の提出をいったん停止することがでできます。
「E2B(R3) 実装ガイドに対応した市販後副作用等報告及び治験副作用等報告について」の一部改正について (wakayama.lg.jp)(令 和 2年8月 31 日 発行)
(イ)開発を長期間中断する場合等
① 開発を長期間中断することが予想される場合、又は申請中において、専門協議後
の照会事項の回答作成に長期間要することが予想される場合は、その旨を機構審査
マネジメント部審査企画課に書面により申し出て、開発が再開されるまで又は照会
事項の回答を提出するまでの間は報告を留保することができる。なお、治験副作用
等報告(研究報告及び外国措置報告を除く。)を留保している期間中にあっても安全
性情報の収集に努め、開発再開時に当該情報を治験薬概要書等及び治験実施計画書
又は申請資料概要へ反映させる。また、開発の再開に伴い副作用等の報告を再開す
る際には必要な書類を機構審査マネジメント部審査企画課に提出すること。
(ただ報告は留保できるけれど、収集に努めるとあり、後に留保解除申請書を提出後に書類に反映させることと記載があります)
他に必要な対応:即時報告、措置報告、安全性定期報告の提出
安全性上の問題で治験中止届を提出する際は、下記記載(6)および(5)に該当し、いずれもの2点で措置報告が必要となる。
Q145:【治験】 外国における措置について、「製造、輸入又は販売の中止、回収、廃棄その他保健衛 生上の危害の発生又は拡大を防止するための措置」に該当するものの例としては、どの ような場合があるか?A145:【治験】 次の場合は外国における措置に該当する。
(1)有効性又は安全性の問題を理由として行われる、効能又は効果、用法及び用量の 変更又は制限
(2)製造、輸入又は販売の中止、及び製造方法の変更等のうち、有効性の不足又は安 全性の問題を理由として行われるもの(例えば、血液製剤でウイルス混入を防ぐ ために不活化工程を導入した場合等)
(3)製品の回収・廃棄のうち、有効性、安全性等の問題を理由としたもの(自主的に 回収した場合を含む)
(4)使用上の注意の改訂(WARNINGS AND PRECAUTIONS 等)のうち、重要な変更 等
(5)治験全体の中断・中止のうち、品質、有効性又は安全性の問題によるもの
(6)治験中のドクターレター等の配布による安全措置の強化等
安全性報告の個別症例は、留保届の提出をもって停止することを書きました。
ただ、その間にも研究報告、措置報告は提出義務があります。
そして安全性定期報告のJ-DSUR/DSURの調査期間に関しては、以下の区切りになります。
- J-DSUR: 前回データロックポイントの翌日~留保解除申出書提出直近の起算日前日まで
- DSUR: 起算日前日までの1年分
ややこしいですね。。。
まとめ
安全性情報の収集や報告については、厳密なタイムラインの管理が必要されます。
薬機法やそれに基づく通知等、規制を遵守し適切な活動を行う必要があるため、通知やタイムライン管理については常にセンシティブである必要があります。
内容の変更は前触れなくいきなり通知されることが多いため
安全性報告の提出義務を適切に履行するには自主的に関連部署と連携をとって
規制がどのような組み立てになっているか、細かく読み解き規制の動きを注視して安全性報告活動をしていかないといけません。
PVは細かい人が多いと言われるのはこのへんですね。
こういう細かい内容について解説しているものがなかなかないので
今回は安全性の収集と提出はいつからいつまですればいいか、そのきっかけとなる当局提出書類について、書いてみました。
自分の備忘録としても書いてみましたが、お役にたてば幸いです。
ではでは。


コメント